
- school Campus Bookshelves
- menu_book Bookshelves
- perm_media Learning Objects
- login Login
- how_to_reg Request Instructor Account
- hub Instructor Commons
- Download Page (PDF)
- Download Full Book (PDF)
- Periodic Table
- Physics Constants
- Scientific Calculator
- Reference & Cite
- Tools expand_more
- Readability
selected template will load here
This action is not available.

Case Study: Cystic Fibrosis - CER
- Last updated
- Save as PDF
- Page ID 26446
This page is a draft and is under active development.
Part I: A Case of Cystic Fibrosis
Dr. Weyland examined a six month old infant that had been admitted to University Hospital earlier in the day. The baby's parents had brought young Zoey to the emergency room because she had been suffering from a chronic cough. In addition, they said that Zoey sometimes would "wheeze" a lot more than they thought was normal for a child with a cold. Upon arriving at the emergency room, the attending pediatrician noted that salt crystals were present on Zoey's skin and called Dr. Weyland, a pediatric pulmonologist. Dr. Weyland suspects that baby Zoey may be suffering from cystic fibrosis.
CF affects more than 30,000 kids and young adults in the United States. It disrupts the normal function of epithelial cells — cells that make up the sweat glands in the skin and that also line passageways inside the lungs, pancreas, and digestive and reproductive systems.
The inherited CF gene directs the body's epithelial cells to produce a defective form of a protein called CFTR (or cystic fibrosis transmembrane conductance regulator) found in cells that line the lungs, digestive tract, sweat glands, and genitourinary system.
When the CFTR protein is defective, epithelial cells can't regulate the way that chloride ions pass across cell membranes. This disrupts the balance of salt and water needed to maintain a normal thin coating of mucus inside the lungs and other passageways. The mucus becomes thick, sticky, and hard to move, and can result in infections from bacterial colonization.

- "Woe to that child which when kissed on the forehead tastes salty. He is bewitched and soon will die" This is an old saying from the eighteenth century and describes one of the symptoms of CF (salty skin). Why do you think babies in the modern age have a better chance of survival than babies in the 18th century?
- What symptoms lead Dr. Weyland to his initial diagnosis?
- Consider the graph of infections, which organism stays relatively constant in numbers over a lifetime. What organism is most likely affecting baby Zoey?
- What do you think is the most dangerous time period for a patient with CF? Justify your answer.
Part II: CF is a disorder of the cell membrane.
Imagine a door with key and combination locks on both sides, back and front. Now imagine trying to unlock that door blind-folded. This is the challenge faced by David Gadsby, Ph.D., who for years struggled to understand the highly intricate and unusual cystic fibrosis chloride channel – a cellular doorway for salt ions that is defective in people with cystic fibrosis.
His findings, reported in a series of three recent papers in the Journal of General Physiology, detail the type and order of molecular events required to open and close the gates of the cystic fibrosis chloride channel, or as scientists call it, the cystic fibrosis transmembrane conductance regulator (CFTR).
Ultimately, the research may have medical applications, though ironically not likely for most cystic fibrosis patients. Because two-thirds of cystic fibrosis patients fail to produce the cystic fibrosis channel altogether, a cure for most is expected to result from research focused on replacing the lost channel.

5. Suggest a molecular fix for a mutated CFTR channel. How would you correct it if you had the ability to tinker with it on a molecular level?
6. Why would treatment that targets the CFTR channel not be effective for 2⁄3 of those with cystic fibrosis?
7. Sweat glands cool the body by releasing perspiration (sweat) from the lower layers of the skin onto the surface. Sodium and chloride (salt) help carry water to the skin's surface and are then reabsorbed into the body. Why does a person with cystic fibrosis have salty tasting skin?
Part III: No cell is an island
Like people, cells need to communicate and interact with their environment to survive. One way they go about this is through pores in their outer membranes, called ion channels, which provide charged ions, such as chloride or potassium, with their own personalized cellular doorways. But, ion channels are not like open doors; instead, they are more like gateways with high-security locks that are opened and closed to carefully control the passage of their respective ions.
In the case of CFTR, chloride ions travel in and out of the cell through the channel’s guarded pore as a means to control the flow of water in and out of cells. In cystic fibrosis patients, this delicate salt/water balance is disturbed, most prominently in the lungs, resulting in thick coats of mucus that eventually spur life-threatening infections. Shown below are several mutations linked to CFTR:

8. Which mutation do you think would be easiest to correct. Justify your answer. 9. Consider what you know about proteins, why does the “folding” of the protein matter?
Part IV: Open sesame
Among the numerous ion channels in cell membranes, there are two principal types: voltage-gated and ligand-gated. Voltage-gated channels are triggered to open and shut their doors by changes in the electric potential difference across the membrane. Ligand-gated channels, in contrast, require a special “key” to unlock their doors, which usually comes in the form of a small molecule.
CFTR is a ligand-gated channel, but it’s an unusual one. Its “key” is ATP, a small molecule that plays a critical role in the storage and release of energy within cells in the body. In addition to binding the ATP, the CFTR channel must snip a phosphate group – one of three “P’s” – off the ATP molecule to function. But when, where and how often this crucial event takes place has remains obscure.

10. Compare the action of the ligand-gated channel to how an enzyme works.
11. Consider the model of the membrane channel, What could go wrong to prevent the channel from opening?
12. Where is ATP generated in the cell? How might ATP production affect the symptoms of cystic fibrosis?
13. Label the image below to show how the ligand-gated channel for CFTR works. Include a summary.

Part V: Can a Drug Treat Zoey’s Condition?
Dr. Weyland confirmed that Zoey does have cystic fibrosis and called the parents in to talk about potential treatments. “Good news, there are two experimental drugs that have shown promise in CF patients. These drugs can help Zoey clear the mucus from his lungs. Unfortunately, the drugs do not work in all cases.” The doctor gave the parents literature about the drugs and asked them to consider signing Zoey up for trials.
The Experimental Drugs
Ivacaftor TM is a potentiator that increases CFTR channel opening time. We know from the cell culture studies that this increases chloride transport by as much as 50% from baseline and restores it closer to what we would expect to observe in wild type CFTR. Basically, the drug increases CFTR activity by unlocking the gate that allows for the normal flow of salt and fluids.
In early trials, 144 patients all of whom were age over the age of 12 were treated with 150 mg of Ivacaftor twice daily. The total length of treatment was 48 weeks. Graph A shows changes in FEV (forced expiratory volume) with individuals using the drug versus a placebo. Graph B shows concentrations of chloride in patient’s sweat.

14. What is FEV? Describe a way that a doctor could take a measurement of FEV.
15. Why do you think it was important to have placebos in both of these studies?
16. Which graph do you think provides the most compelling evidence for the effectiveness of Ivacafor? Defend your choice.
17. Take a look at the mutations that can occur in the cell membrane proteins from Part III. For which mutation do you think Ivacaftor will be most effective? Justify your answer.
18. Would you sign Zoey up for clinical trials based on the evidence? What concerns would a parent have before considering an experimental drug?
Part VI: Zoey’s Mutation
Dr. Weyland calls a week later to inform the parents that genetic tests show that Zoey chromosomes show that she has two copies of the F508del mutation. This mutation, while the most common type of CF mutation, is also one that is difficult to treat with just Ivacaftor. There are still some options for treatment.
In people with the most common CF mutation, F508del, a series of problems prevents the CFTR protein from taking its correct shape and reaching its proper place on the cell surface. The cell recognizes the protein as not normal and targets it for degradation before it makes it to the cell surface. In order to treat this problem, we need to do two things: first, an agent to get the protein to the surface, and then ivacaftor (VX-770) to open up the channel and increase chloride transport. VX-809 has been identified as a way to help with the trafficking of the protein to the cell surface. When added VX-809 is added to ivacaftor (now called Lumacaftor,) the protein gets to the surface and also increases in chloride transport by increasing channel opening time.

In early trials, experiments were done in-vitro, where studies were done on cell cultures to see if the drugs would affect the proteins made by the cell. General observations can be made from the cells, but drugs may not work on an individual’s phenotype. A new type of research uses ex-vivo experiments, where rectal organoids (mini-guts) were grown from rectal biopsies of the patient that would be treated with the drug. Ex-vivo experiments are personalized medicine, each person may have different correctors and potentiators evaluated using their own rectal organoids. The graph below shows how each drug works for 8 different patients (#1-#8)
19. Compare ex-vivo trials to in-vitro trials.
20. One the graph, label the group that represents Ivacaftor and Lumacaftor. What is the difference between these two drugs?
21. Complete a CER Chart. If the profile labeled #7 is Zoey, rank the possible drug treatments in order of their effectiveness for her mutation. This is your CLAIM. Provide EVIDENCE to support your claim. Provide REASONING that explains why this treatment would be more effective than other treatments and why what works for Zoey may not work for other patients. This is where you tie the graph above to everything you have learned in this case. Attach a page.
A Case of Cystic Fibrosis (KEY)

What educators are saying
Description.
This is the answer key for the case study on cystic fibrosis where students explore how children are diagnosed with CF, how CF mutations affect transport across the cell membrane, and how two drugs can be used to treat the disease. The activity is used in AP Biology class and requires students to complete a CER (claim, evidence, reasoning) at the end to justify the best course of treatment for patient Zoey.
The student version of this case is available for free at biologycorner.com and includes two versions, in-person and remote learning version.
Download provides the answer key and links to the Google docs for the student worksheets for both in-person and remote learning. Document also includes rubric for CER and additional resources for teaching.
Questions & Answers
Biologycorner.
- We're hiring
- Help & FAQ
- Privacy policy
- Student privacy
- Terms of service
- Tell us what you think

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Respir Med Case Rep
Case report: Three adult brothers with cystic fibrosis (delF508-delF508) maintain unusually preserved clinical profile in the absence of standard CF care
We present three cases in this report. Three adult brothers, homozygous for the delF508 cystic fibrosis mutation, have maintained an unusually preserved clinical condition even though they did not attend a CF Clinic during their childhood, do not attend a CF Clinic now, and do not follow standard CF care guidelines. The brothers use an alternative CF treatment regimen on which they have maintained normal lung function, height/weight, and bloodwork, and they utilize less than half the recommended dosage of pancreatic enzymes. The brothers culture only methicillin-sensitive Staphylococcus aureus, and have never cultured any other bacteria. Highly effective modulator therapies, such as elexacaftor/tezacaftor/ivacaftor, do not substantially reduce infection and inflammation in vivo in CF patients, and thus these three case reports are of special note in terms of suggesting adjunct therapeutic approaches. Finally, these three cases also raise important questions about standard CF care guidelines.
- • Three adult brothers, delF508 cystic fibrosis (CF) homozygotes, maintain unusually preserved clinical condition absent standard CF care.
- • An alternative CF treatment regimen has kept their lung function, weight/height, and lab parameters normal, with low pancreatic enzyme dose.
- • The brothers culture only methicillin-sensitive Staphylococcus aureus, and have never cultured any other bacteria.
- • Highly effective modulator therapies (HEMT) for CF do not substantially reduce infection and inflammation in vivo; these cases are thus of note.
- • These cases also raise important questions about standard CF care guidelines.
1. Introduction
Cystic fibrosis (CF) is a serious and life-shortening genetic disorder affecting approximately 70,000 persons worldwide [ 1 ]. Respiratory failure is the foremost cause of death in CF patients, and lung transplantation is often considered in end-stage CF disease. For those born with CF in the last five years, median predicted survival age is now 44, which is decades longer than survival rates in the recent past [ 2 ]. Indeed, new advances in CF modulator therapy and CF gene therapy may eventually provide a normal life expectancy for these individuals.
A key approach in fighting the ravages of CF while waiting for more advanced treatments to be developed has been to slow the inexorable decline in lung function. Typical rate of lung function decline in CF is approximately −1.2 to −1.6 FEV1% per year [ 3 ]. Rate of decline is strongly associated with type of CF mutation. The three most severe classes of CFTR, Classes I, II, and III, represent defects in protein production, protein processing, and protein regulation, respectively [ 4 ]. The most common CF-causing mutation is delF508, occurring in 70% of cases, which is a Class II mutation [ 5 ]. Being homozygous for the delF508 mutation confers a severe phenotype, including pancreatic insufficiency and a steeper rate of decline in lung function over time [ 6 ]. In the United States, it is estimated that approximately 50% of those with cystic fibrosis are homozygous for delF508 [ 7 ]. Standard clinical care for severe mutation cases is often aggressive, including but not limited to daily airway clearance, use of pancreatic enzymes at the level of 500-2,500 lipase units/kg/meal (and enteric feeding if adequate weight percentile cannot be maintained), common and repeated use of oral, inhaled and intravenous antibiotics, daily intake of water-miscible versions of fat-soluble vitamins, and quarterly CF Clinic visits where lung function parameters and cultures of lung bacteria and fungi are assessed [ 8 , 9 ]. Pulmonary exacerbations often result in hospitalization, which may occur one or more times per year, typically lasting 14–21 days and including intensive antibiotic treatment and chest physical therapy. Everyday treatment burden is high, with estimates of 2–3 hours per day, with adherence at an estimated 50% or less [ 10 ]. The mean annual cost of standard supportive CF care in the US in 2016 (in 2019 dollars), before CFTR modulator therapies, was estimated to average $77,143, with severe non-transplant cases experiencing multiple pulmonary exacerbations costing on average triple or quadruple that amount [ 11 ]. With the average cost of elexacaftor/tezacaftor/ivacaftor (Trikafta) treatment currently over $311,000 per year, average standard supportive CF care costs were expected to double in 2019 [ 12 ] and increase further over time, perhaps quadrupling, with wider adoption of that treatment by all eligible patients.
Here we report on three adult brothers who are delF508 homozygotes, and yet who have maintained an unusually preserved clinical profile in the absence of standard CF clinical care. At the time of this writing, Brother A is 23 years old, Brother B is 21 years old, and Brother C is 18 years old. They are full-blooded siblings.
2. Case reports
2.1. brother a.
Brother A, now aged 23, was born full-term weighing 10 lbs. 2 oz. to a carrier mother experiencing gestational diabetes who subsequently breastfed him. His weight percentile decreased significantly over time, and at 6 months, after a course of oral antibiotics for a suspected ear infection, he developed a severe Vitamin K deficiency manifesting in quarter-sized black bruises on his body, as well as Pseudo-Bartter Syndrome. He was hospitalized until IV fluids stabilized his condition and normalized his electrolytes. Vitamin K shots were also administered. At 9 months of age, he was diagnosed with cystic fibrosis, and the genetic mutation analysis identified him as a delF508 homozygote. Between the time of his hospitalization and his diagnosis, he suffered from malnutrition with accompanying protein edema and his weight percentile, which had been over 97th percentile when born, was under the 5th percentile adjusted for age and sex. Once started on pancreatic enzymes (CREON 5) after diagnosis, his weight percentile increased to approximately the 30th percentile.
Approximately one year after diagnosis, the parents of Brother A elected to depart from standard CF care, including an election to stop attending the CF Clinic, while continuing to be under the care of their family pediatrician. The treatment plan for the brothers is described in detail in a later section. The only prescription medicine taken during his childhood and continuing to this day remains CREON 5/6, with Brother A utilizing 4 CREON 5/6 per meal, less than half the lowest recommended dose for his weight. In the teen years, Brother A experienced three episodes of heat exhaustion requiring IV fluid stabilization in an emergency room, has had one endoscopic sinus cleaning for sinus pain at age 20, and also underwent an appendectomy for appendicitis at age 23, but otherwise has had no major clinical issues, though exhibiting digital clubbing. Brother A played ice hockey throughout his childhood and teen years. His height, weight, lung function, and lab results at age 23 are provided in Table 1 .
Clinical parameters, Brother A.
2.2. Brother B
Brother B, now aged 21, was born full-term, weighing 8 lbs. 8 oz., the mother supplementing with oral glutathione (GSH) during the pregnancy and subsequently breastfeeding him. Brother B has never attended a CF Clinic, was diagnosed at 2 weeks of age, and was under the care of the family's pediatrician only. Brother B's only prescription medication during his childhood was CREON 5/6, just as with Brother A, utilizing 4 capsules per meal. Brother B has never needed to be hospitalized or have surgery or antibiotics. While Brother B does not exhibit digital clubbing; when recovering from respiratory viruses, he does manifest a cough that lingers longer than it lingers for his brothers, though the cough ultimately resolves. Brother B played ice hockey in childhood and teen years, as well as participated in gymnastics, cross-country running, track and field, and weight-lifting. His height, weight, lung function, and lab results at age 21 are provided in Table 2 .
Clinical parameters, Brother B.
2.3. Brother C
Brother C, now aged 18, was born full-term weighing 9 lbs. 2 oz., the mother supplementing with oral glutathione (GSH) during the pregnancy and subsequently breastfeeding him. Brother C has never attended a CF Clinic, was diagnosed at 2 weeks of age, and was under the care of the family's pediatrician only. Brother C's only prescription medication during his childhood was CREON 5/6, just as with Brothers A and B, utilizing 4 capsules per meal. Brother C has never needed to be hospitalized, or have surgery or antibiotics. Brother C does not exhibit digital clubbing. Brother C played ice hockey in childhood and teen years, as well as participated in gymnastics. His height, weight, lung function, and lab results at age 18 are provided in Table 3 .
Clinical parameters, Brother C.
3. Description of treatment
Given the severity of the genotype involved and the almost complete non-adherence to standard CF guidelines (with the exception of a significantly lower-than-average dose of prescription pancreatic enzymes and a standard dose of water-miscible fat soluble vitamins), the preserved clinical profile of these three brothers is noteworthy. However, the family developed a regimen that went well beyond pancreatic enzymes and water-miscible vitamins. The treatment regimen is provided in Table 4 .
Description of Daily Regimen.

4. Discussion
There are several possibilities for the preserved clinical status of these three brothers in the absence of standard CF care:
- a) They avoided the CF Clinic setting. Recent research [ 13 ] has shown that Pseudomonas infections are more prevalent and lung function lower among CF patients in standard care versus CF patients in a telemedicine setting. It is possible these three brothers benefitted from not attending a standard CF Clinic, especially since during their childhood years at the turn of the century, Clinic infection control was not emphasized. For example, during Brother A's first few CF Clinic visits as an infant, families were expected to wait together in a communal area with communal toys, and health care professionals at the Clinic wore neither masks nor gloves as they moved from exam room to exam room.
- b) With the exception of Brother A, Brothers B and C have used no antibiotics at all. Brother A has only used antibiotics three times in his life; the first use in infancy precipitated Pseudo-Bartter Syndrome, leading to his diagnosis with cystic fibrosis. The other two uses were incident to endoscopic sinus scraping and an appendectomy. Recent research has shown the importance of the gut microbiome in maintenance of health (including respiratory function), digestion and immune signaling, and this is true in the case of cystic fibrosis as well [ [14] , [15] , [16] ]. As David Pride, Associate Director of Microbiology at UC San Diego, notes in an address to the 2019 North American Cystic Fibrosis Conference [ 17 ], “It is important to preserve our microbiomes because they play important roles in preventing pathogens from establishing infections, in the development of our immune systems to recognize and kill pathogens, and in metabolic processes such as the digestion of foods. Indiscriminate uses of antibiotics can have profound and long-lasting effects upon our microbiomes by killing many of the bacteria that make up our microbiome; thus, limiting their use may aid in keeping us healthy.”
- Prevalent, sometimes chronic, antibiotic use among CF patients results in a significant gut dysbiosis [ 18 ]. In addition, it has been noted that aggressive antibiotic use in CF, usually incident to the first manifestation of Staphylococcus aureus (SA), may allow Pseudomonas aeruginosa a greater foothold [ 19 ], and that aggressive treatment of Pseudomonas may, in turn, promote drug resistance and may allow additional bacteria, such as Stenotrophomonas maltophilia, an opportunity to proliferate [ 20 ]. Perhaps a preserved gut microbiome due to non-use of antibiotics may have played a role in the brothers' preserved clinical condition; this may also help account for the brothers’ significantly lower level of need for pancreatic enzymes. Perhaps also the decision not to aggressively treat their light to moderate growth of methicillin-sensitive SA may have precluded additional bacteria, including drug-resistant bacteria, from emerging.
- c) Other standard daily CF treatments were not employed, either, which might help account for their preserved clinical condition. For example, the brothers do not use bronchodilators; and beta-2 agonist bronchodilators, such as albuterol, have recently been shown to significantly reduce delF508 CFTR activation [ 21 ]. This reduction is even evident when CFTR modulators are used, with the finding of a more than 60% reduction of modulator-corrected CFTR activation in vitro, “sufficient to abrogate VX809/VX770 modulation of F508-del CFTR” [ 21 ]. In addition, the brothers do not use DNase, which has been associated with increased levels of neutrophil elastase in past research [ 22 ]. Last, after Brother A transitioned to his new treatment regimen at approximately 23 months of age, chest percussive therapy (CPT) was discontinued, and neither Brother B nor C underwent CPT at all. A Cochrane meta-review found that while CPT constituted the lion's share of treatment time burden in CF, the evidence that outcomes of CPT differed from no CPT was “very low quality” [ 23 ].
- d) Glutathione (GSH) is heavily emphasized in the brothers' daily regimen. Levels of GSH are strongly decreased in the extracellular milieu of CF patients, as its efflux from epithelial cells is compromised by CFTR mutation [ 24 ]. In the non-CF research literature, GSH in its ratio of reduced to oxidized forms (GSH:GSSG) has been shown to be the foundation of redox signaling in the body; GSH is also the body's primary water-soluble antioxidant and a potent mucolytic, and conserves NO through formation of GSNO. Given its pivotal roles, it is not surprising to find that GSH deficiency is noted in several other severe respiratory illnesses besides CF, including ARDS, COPD, IIP, IPF, IRDS, and DFA, and GSH deficiency is a key catalyst for (and GSH dosing a key treatment of) cachexia [ 24 ]. The use of GSH in the treatment of CF may reduce systemic inflammation, lessen the viscosity of mucus, and catalyze the efficacy of the immune system, including through GSNO. Indeed, a clinical study by Visca et al. found significantly increased BMI [ 25 ], significantly increased lung function [ 26 ], and even improved bacteriological results [ 27 ] from the daily use of oral glutathione in children with CF at a dose of 30 mg/lb body weight/day, spread out over 3–4 doses, over a time period of 6 months. In addition, the parents of these brothers noted a sudden increase in both saliva and appetite in Brother A after glutathione (GSH) was introduced when he was two years of age. Brothers B and C, on GSH from two weeks of birth (and with the mother supplementing with oral glutathione throughout pregnancy with these two brothers), never displayed low saliva or low appetite. The preserved clinical status of these three brothers may perhaps be related to this glutathione-heavy regimen.
- e) Other aspects of the brothers' regimen may offset their disease condition. The use of probiotics [ 28 ], the heavy emphasis on antioxidants in addition to glutathione (such as C, CoQ10, Alpha-lipoic acid, D, E, etc. [ 29 ]), amino acids (such as cysteine [ 30 ], carnitine [ 31 ], choline [ 32 , 33 ], taurine [ 34 ], and glycine [ 35 ]), curcumin [ 36 ], and additional digestive support beyond enzymes (lecithin, bile acid). It is possible that some or all of these supplementation efforts also helped to preserve the clinical status of the three brothers. In addition, exclusive breastfeeding of CF infants has been linked to significantly higher FEV1 at age 5 (difference significant at p ≤ 0.001 between breastfed and formula fed CF infants), perhaps contributing to the preservation of lung function beyond that time frame [ 37 ].
- f) Modifier alleles may be present. While no in-depth analysis of the brothers' genetic profile has been performed beyond the identification of their CF mutations, there are known modifier alleles that serve to lessen (or exacerbate) the severity of CF (see, for example [ 38 ]). It is possible all three brothers inherited some propitious set of modifier alleles.
5. Conclusion
In conclusion, while it is encouraging and heartening that new CF therapies, such as elexacaftor/tezacaftor/ivacaftor (Trikafta) and other HEMT (highly effective modulator therapies), now exist, it is instructive to consider how this family was able to preserve the clinical condition of three brothers, all delF508 homozygotes, in the absence of those therapies, and even in the absence of standard CF care. While HEMT certainly increase CFTR activity, there is substantially less effect on infection and inflammation in vivo [ 39 ]. As recently noted by Singh et al., “[I]f infection and inflammation become uncoupled from CFTR activity in established disease [due to HEMT use], drugs targeting CFTR may need to be initiated very early in life, or used in combination with agents that suppress infection and inflammation ” [ 39 ; emphasis ours]. These case reports may speak to that proposition.
Furthermore, each possible explanation for that preservation is an occasion for reflection on the current standard of CF care. We may feel to ask questions such as, “From the point of view of the patient's health, is the entire concept of the CF Clinic inherently flawed? Is the frequent, sometimes chronic, use of antibiotics and certain other medications in CF care a real double-edged sword for CF patients, with disadvantages possibly outweighing advantages in many cases? Are there measures we can take now, relatively inexpensive measures such as the use of glutathione (GSH) and other antioxidants and amino acids, that will help preserve the clinical status of CF patients, and that might synergize with cutting-edge treatments such as CFTR modulators to improve and safeguard health to an even greater degree, and which should be initiated as early in life as possible, possibly while the fetus is still in utero ?” The experience of these three brothers, so removed from standard CF care and yet so well preserved in their clinical status, highlights the need to consider such questions more urgently than we perhaps have heretofore considered them.
Funding sources
This work was supported by the Utah Valley Institute of Cystic Fibrosis, for publication costs only.
Acknowledgements
The author wishes to acknowledge Valerie M. Hudson, who assisted with the writing of this article.
- Publications
- Conferences & Events
- Professional Learning
- Science Standards
- Awards & Competitions
- Daily Do Lesson Plans
- Free Resources
- American Rescue Plan
- For Preservice Teachers
- NCCSTS Case Collection
- Partner Jobs in Education
- Interactive eBooks+
- Digital Catalog
- Regional Product Representatives
- e-Newsletters
- Bestselling Books
- Latest Books
- Popular Book Series
- Prospective Authors
- Web Seminars
- Exhibits & Sponsorship
- Conference Reviewers
- National Conference • Denver 24
- Leaders Institute 2024
- National Conference • New Orleans 24
- Submit a Proposal
- Latest Resources
- Professional Learning Units & Courses
- For Districts
- Online Course Providers
- Schools & Districts
- College Professors & Students
- The Standards
- Teachers and Admin
- eCYBERMISSION
- Toshiba/NSTA ExploraVision
- Junior Science & Humanities Symposium
- Teaching Awards
- Climate Change
- Earth & Space Science
- New Science Teachers
- Early Childhood
- Middle School
- High School
- Postsecondary
- Informal Education
- Journal Articles
- Lesson Plans
- e-newsletters
- Science & Children
- Science Scope
- The Science Teacher
- Journal of College Sci. Teaching
- Connected Science Learning
- NSTA Reports
- Next-Gen Navigator
- Science Update
- Teacher Tip Tuesday
- Trans. Sci. Learning
MyNSTA Community
- My Collections
Maggie’s Illness
Protein Structure and Function in Cystic Fibrosis
By Michaela Gazdik Stofer
Share Start a Discussion

This directed case study examines the molecular basis of cystic fibrosis to emphasize the relationship between the genetic code stored in a DNA sequence and the encoded protein’s structure and function. Cystic fibrosis is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) protein that functions to help maintain salt and water balance along the surface of the lung and gastrointestinal tract. This case introduces students to “Maggie,” who has just been diagnosed with cystic fibrosis. The students must identify the mutation causing Maggie’s disease by transcribing and translating a portion of the wildtype and mutated CFTR gene. Students then compare the three-dimensional structures of the resulting proteins to better understand the effect a single amino acid mutation can have on the overall shape of a protein. Students also review the concepts of tonicity and osmosis to examine how the defective CFTR protein leads to an increase in the viscosity of mucus in cystic fibrosis patients. This case was developed for use in an introductory college-level biology course but could also be adapted for use in an upper-level cell or molecular biology course.
Download Case
Date Posted
- Generate a protein sequence through transcription and translation of a given DNA gene sequence.
- Explain the chemistry of amino acid side chains and their importance in protein folding.
- Describe how a mutation in a protein sequence leads to changes in the overall tertiary structure of the protein.
- Examine various levels of protein structure using Cn3D to view three-dimensional protein structures from NCBI’s Entrez Structure database.
- Relate the loss of function of the CFTR protein to the physiological causes of cystic fibrosis.
Protein structure; transcription; translation; DNA mutation; cystic fibrosis; genetic disease; protein function; protein folding; protein; CFTR; Cn3D
Subject Headings
EDUCATIONAL LEVEL
Undergraduate lower division, Undergraduate upper division
TOPICAL AREAS
TYPE/METHODS
Teaching Notes & Answer Key
Teaching notes.
Case teaching notes are protected and access to them is limited to paid subscribed instructors. To become a paid subscriber, purchase a subscription here .
Teaching notes are intended to help teachers select and adopt a case. They typically include a summary of the case, teaching objectives, information about the intended audience, details about how the case may be taught, and a list of references and resources.
Download Notes
Answer Keys are protected and access to them is limited to paid subscribed instructors. To become a paid subscriber, purchase a subscription here .
Download Answer Key
Materials & Media
Supplemental materials.
The following two files should be viewed with the Cn3D software to view a single domain of the CFTR and ∆F508 CFTR proteins.
You may also like
Web Seminar
Join us on Thursday, June 13, 2024, from 7:00 PM to 8:00 PM ET, to learn about the science and technology of firefighting. Wildfires have become an e...
Join us on Thursday, October 10, 2024, from 7:00 to 8:00 PM ET, for a Science Update web seminar presented by NOAA about climate science and marine sa...
Secondary Pre-service Teachers! Join us on Monday, October 21, 2024, from 7:00 to 8:15 PM ET to learn about safety considerations for the science labo...
Elementary Pre-service Teachers! Join us on Monday, October 7, 2024, from 7:00 – 8:15 PM ET to learn about safety considerations for the element...
Case Study - What is the Relationship Between the Cell Membrane and Cystic Fibrosis?
CF affects more than 30,000 kids and young adults in the United States. It disrupts the normal function of epithelial cells — cells that make up the sweat glands in the skin and that also line passageways inside the lungs, pancreas, and digestive and reproductive systems.
The inherited CF gene directs the body's epithelial cells to produce a defective form of a protein called CFTR (or cystic fibrosis transmembrane conductance regulator) found in cells that line the lungs, digestive tract, sweat glands, and genitourinary system.
When the CFTR protein is defective, epithelial cells can't regulate the way that chloride ions pass across cell membranes. This disrupts the balance of salt and water needed to maintain a normal thin coating of mucus inside the lungs and other passageways. The mucus becomes thick, sticky, and hard to move, and can result in infections from bacterial colonization.

1. "Woe to that child which when kissed on the forehead tastes salty. He is bewitched and soon will die"
This is an old saying from the eighteenth century and describes one of the symptoms of CF (salty skin). Why do you think babies in the modern age have a better chance of survival than babies in the 18th century?
2. What symptoms lead Dr. Weyland to his initial diagnosis?
3. Consider the graph of infections, which organism stays relatively constant in numbers over a lifetime?
What organism is most likely affecting baby Zoey?
4. Explain how the CF gene affects the cell membrane.
5. Consider what you know about TONICITY and the cell membrane. Why is it important to regulate salt in cells?
Part II: CF is a disorder of the cell membrane.
Imagine a door with key and combination locks on both sides, back and front. Now imagine trying to unlock that door blind-folded. This is the challenge faced by David Gadsby, Ph.D., who for years struggled to understand the highly intricate and unusual cystic fibrosis chloride channel – a cellular doorway for salt ions that is defective in people with cystic fibrosis.
His findings, reported in a series of three recent papers in the Journal of General Physiology, detail the type and order of molecular events required to open and close the gates of the cystic fibrosis chloride channel, or as scientists call it, the cystic fibrosis transmembrane conductance regulator (CFTR).
Ultimately, the research may have medical applications, though ironically not likely for most cystic fibrosis patients. Because two-thirds of cystic fibrosis patients fail to produce the cystic fibrosis channel altogether, a cure for most is expected to result from research focused on replacing the lost channel.

6. Compare the normal and the mutant CFTR protein. How would you correct the mutant protein if you had the ability to tinker with it on a molecular level?
7. Why would treatment that targets the CFTR channel not be effective for ⅔ of those with cystic fibrosis? 8. Sweat glands cool the body by releasing perspiration (sweat) from the lower layers of the skin onto the surface. Sodium and chloride (salt) help carry water to the skin's surface and are then reabsorbed into the body. Why does a person with cystic fibrosis have salty tasting skin?
Part III: No cell is an island
Like people, cells need to communicate and interact with their environment to survive. One way they go about this is through pores in their outer membranes, called ion channels, which provide charged ions, such as chloride or potassium, with their own personalized cellular doorways. But, ion channels are not like open doors; instead, they are more like gateways with high-security locks that are opened and closed to carefully control the passage of their respective ions.

9. Which mutation do you think would be easiest to correct? Justify your answer.
10. Consider what you know about proteins, why does the "folding" of the protein matter?
Part IV: Open Sesame

Among the numerous ion channels in cell membranes, there are two principal types: voltage-gated and ligand-gated. Voltage-gated channels are triggered to open and shut their doors by changes in the electric potential difference across the membrane. Ligand-gated channels, in contrast, require a special “key” to unlock their doors, which usually comes in the form of a small molecule.
CFTR is a ligand-gated channel, but it’s an unusual one. Its “key” is ATP, a small molecule that plays a critical role in the storage and release of energy within cells in the body. In addition to binding the ATP, the CFTR channel must snip a phosphate group – one of three “P’s” – off the ATP molecule to function. But when, where and how often this crucial event takes place has remained obscure.
11. Label the image to the right to show how the ligand-gated channel for CFTR works. (Structures: Ligand-gated channel protein, ATP, phospholipids). Summarize how this channels works.
12. Where is ATP generated in the cell? How might ATP production affect the symptoms of cystic fibrosis?
Part V: Can a Drug Treat Zoey's Condition?
Dr. Weyland confirmed that Zoey does have cystic fibrosis and called the parents in to talk about potential treatments. “Good news, there are two experimental drugs that have shown promise in CF patients. These drugs can help Zoey clear the mucus from her lungs. Unfortunately, the drugs do not work in all cases.” The doctor gave the parents literature about the drugs and asked them to consider signing Zoey up for trials.
The Experimental Drugs
Ivacaftor ™ is a potentiator that increases CFTR channel opening time. We know from the cell culture studies that this increases chloride transport by as much as 50% from baseline and restores it closer to what we would expect to observe in wild type CFTR. Basically, the drug increases CFTR activity by unlocking the gate that allows for the normal flow of salt and fluids.
In early trials, 144 patients all of whom were over the age of 12 were treated with 150 mg of Ivacaftor twice daily. The total length of treatment was 48 weeks. Graph A shows changes in FEV (forced expiratory volume) with individuals using the drug versus a placebo. Graph B shows concentrations of chloride in patient’s sweat.

13. What is FEV (if you're not sure, look this one up)? Describe a way that a doctor could take a measurement of FEV.
14. Why do you think it was important to have placebos in both of these studies?
15. Which graph do you think provides the most compelling evidence for the effectiveness of Ivacaftor. Defend your choice.
16. Take a look at the mutations that can occur in the cell membrane protein from Part III. For which mutation do you think Ivacaftor will be most effective. Justify your answer.
17. Would you sign Zoey up for clinical trials based on the evidence? What concerns would a parent have before considering an experimental drug?
Part VI: Zoey's Mutation
Dr. Weyland calls a week later to inform the parents that genetic tests show that Zoey chromosomes show that she has two copies of the F508del mutation. This mutation, while the most common type of CF mutation, is also one that is difficult to treat with just Ivacaftor. There are still some options for treatment.
In people with the most common CF mutation, F508del, a series of problems prevents the CFTR protein from taking its correct shape and reaching its proper place on the cell surface. The cell recognizes the protein as not normal and targets it for degradation before it makes it to the cell surface. In order to treat this problem, we need to do two things: first, an agent to get the protein to the surface, and then ivacaftor (VX-770) to open up the channel and increase chloride transport. VX-809 has been identified as a way to help with the trafficking of the protein to the cell surface. When added VX-809 is added to ivacaftor (now called Lumacaftor,) the protein gets to the surface and also increases in chloride transport by increasing channel opening time.
In early trials, experiments were done in-vitro, where studies were done on cell cultures to see if the drugs would affect the proteins made by the cell. General observations can be made from the cells, but drugs may not work on an individual’s phenotype. A new type of research uses ex-vivo experiments, where rectal organoids (mini-guts) were grown from rectal biopsies of the patient that would be treated with the drug. Ex-vivo experiments are personalized medicine, each person may have different correctors and potentiators evaluated using their own rectal organoids.
The graph below shows how each drug works for 8 different patients (#1-#8). Swelling in the organoid indicates the the channels within the cell membrane are allowing material to pass.

19. . Compare ex-vivo trials to in-vitro trials.
20. One the graph, label the group that represents Ivacaftor and Lumacaftor. What is the difference between these two drugs?
21. Complete a CER Chart.
If the profile labeled #7 is Zoey, rank the possible drug treatments in order of their effectiveness for her mutation. This is your CLAIM. Provide EVIDENCE to support your claim Provide REASONING that explains why this treatment would be more effective than other treatments and why what works for Zoey may not work for other patients. This is where you tie the graph above to everything you have learned in this case. Attach a page.

Source & Credits
- "CFTR Protein Panels" by Lbudd14 - Own work. Licensed under Creative Commons Attribution-Share Alike 3.0 via Wikimedia Commons
- http://newswire.rockefeller.edu/2003/12/19/scientists-finally-pry-stubborn-cellular-door-ajar/
- http://en.wikipedia.org/wiki/Cystic_fibrosis
- http://www.medscape.org/viewarticle/806649_transcript
- http://www.cff.org/research/clinicalresearch/faqs/combinedkalydeco-vx-809/#Expanded-Access
- Ifacaftor Trial Graph: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3230303/
- Organoid swelling graph: http://www.potentiate.info/?q=trio-clinical-trial-ivacaftor-genistein
- Life expectancy graph: http://www.nationaljewish.org/healthinfo/conditions/cysticfibrosis/life-expectancy/
Follow-up Article: What it's like to have two kids with cystic fibrosis More information at John Hopkins Cystic Fibrosis Center
Other Resources on Cystic Fibrosis
Cystic Fibrosis Mutations Cell Membrane and Transport (Slides)
Molecular Biology Simulations for Case-Based Learning in Biology
Key to cystic fibrosis cases.
M utation/techniques illustrated
For Case A : The DNA sequence files are from the human MP6d9 gene, GenBank accession #M37523, 383 bp, which is linked to the cystic fibrosis gene on chromosome 7. This simulates a PCR-amplified region near the CF gene. The mutated sequence files were created by replacing the G to A base change at nucleotide 170 which causes the loss of an Msp I site. No Southern blotting is necessary to observe the mutation. RFLP marker outside of the actual disease gene, but genetically linked to the disease gene. Reference: Huth,A., et al., Nucleic Acids Res. 17 (17), 7118, 1989. For Case B : The DNA sequences are from the human CFTR gene, GenBank accession #M28668, 6129 bp. The mutated sequence files were created by deleting nucleotides 1653-1655 (delTTC). The normal probe is 25 bp spanning this region, while the CF mutation probe contains the TTC deletion. Reference: Riordan et al., Science 245:1066-1073, 1989.
Case A :
As Sharon Brown browsed the local newspaper, she noticed the story about the five-year old boy with cystic fibrosis who lives on the next block. The article was mainly a human interest story about how the family was coping. There also was some background information about the disease and its inheritance patterns, including the statistics indicating that approximately 1 in 18 people in this part of Minnesota carried a cystic fibrosis mutation. Sharon is two months pregnant. She realizes that she and her husband, Bob, should have been tested for the cystic fibrosis (CF) mutation since they each have some family history of the disease, but they really hadn’t expected to have a child so soon. She discusses this with her physician during her check-up the next day, and together they decide to test Sharon and Bob for a mutation in linked to the CF gene. They also decide to test the developing fetus. Two other families in the same town who also are in the first trimester of a pregnancy, Jill and Mike Jones and Carol and Ron Smith, also decide to be tested after reading the article.
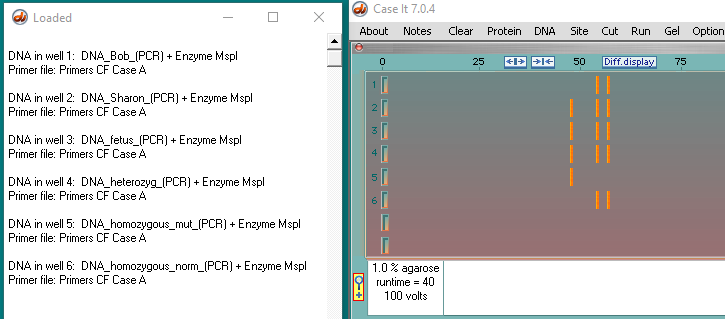
- Brown family:
- Bob – homozygous normal
- Sharon- heterozygous
- Fetus – heterozygous
- Jones family :
- Jill – heterozygous
- Mike – heterozygous
- fetus – homozygous CF mutation
- Smith family :
- Carol – heterozygous
- Ron – heterozygous
- Fetus – homozygous normal
- Controls on all three gels :
- DNA heterozygous control
- DNA homozygous mutation control
- DNA homozygous normal control
Case B : (Contributed by Stephanie Dahlby, Dan Tally, and Janelle Veerkamp, students at UW-River Falls)
Lynda and Jim are expecting their first child. Recently, however, they learn that Lynda’s aunt died of CF and Jim’s uncle died of CF. They are worried that they might be carriers for the disease and pass cystic fibrosis on to their unborn child. They learn about a procedure which can determine whether they are carriers. They also learn about a procedure called amniocentesis which can detect if their unborn child has CF or is a carrier. However, amniocentesis is a very risky procedure. Jim and Lynda ultimately decide that they first want to be tested to see if they are carriers for the disease. If they learn that they both are carriers, they would like to go through with the amniocentesis to see if their child is affected.
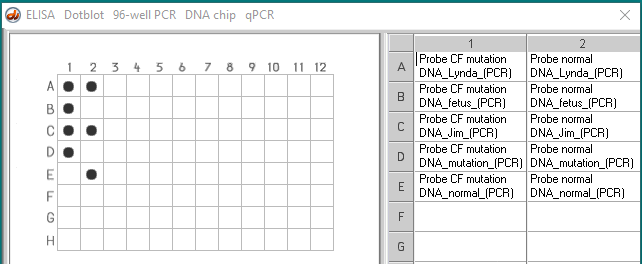
- Lynda – heterozygous
- Fetus – homozygous CF mutation
- Jim – heterozygous
- Control DNA with F508 mutation
- Control normal DNA, without mutation
Case C :
The pre-marriage counseling session Carl and Maggie are having with Pastor Frank is not going at all as they had expected it to. After some of the anticipated discussion of relationship issues, the conversation turns to family planning. When both Carl and Maggie say they want to have children, Pastor Frank, instead of giving advise on how to properly rear children, begins to talk about genetic testing for Cystic Fibrosis! It turns out that Pastor Frank and his wife had two children affected with CF who died in their early teens. Because of the relatively high frequency of CF carriers and his opposition to abortion, Pastor Frank believes that all couples should be tested for the CF gene before getting married. Carl and Maggie are not sure they share Pastor Frank’s beliefs but decide to go along with being tested.
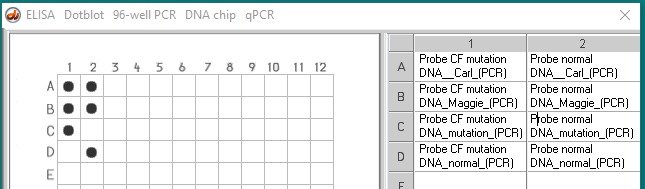
- Carl – heterozygous
- Maggie – heterozygous
- Control normal DNA without mutation
IMAGES
VIDEO
COMMENTS
The inherited CF gene directs the body's epithelial cells to produce a defective form of a protein called CFTR (or cystic fibrosis transmembrane conductance regulator) found in cells that line the lungs, digestive tract, sweat glands, and genitourinary system. When the CFTR protein is defective, epithelial cells can't regulate the way that ...
HESI Cystic fibrosis Case Study Darla. Teacher 25 terms. Debbie_Rowan. Preview. Holistic Nursing Quiz 2. 7 terms. nicole_eveland6. Preview. Exam 1 nursing 3. 179 terms. sandrati2. Preview. HESI RN Case Study - Cystic Fibrosis Peds. 32 terms. ABMan8612. Preview. HESI: Cystic Fibrosis and Rationale. 26 terms. K_Copeland5. Preview. N434 Exam 3 ...
Justify your answer. Part II: CF is a disorder of the cell membrane. Imagine a door with key and combination locks on both sides, back and front. Now imagine trying to unlock that door blind-folded. This is the challenge faced by David Gadsby, Ph.D., who for years struggled to understand the highly intricate and unusual cystic fibrosis chloride ...
This is the answer key for the case study on cystic fibrosis where students explore how children are diagnosed with CF, how CF mutations affect transport across the cell membrane, and how two drugs can be used to treat the disease. The activity is used in AP Biology class and requires students to complete a CER (claim, evidence, reasoning) at ...
The nurse educator obtained a contact number for a CF clinic near the beach in case an exacerbation occurs. Pamela and Donald now feel confident as they plan their first vacation with Darla. Study with Quizlet and memorize flashcards containing terms like Meet the Client: Darla WilliamsPamela and Donald Williams bring their 24-month-old ...
CF key genomics case study key cystic fibrosis exemplar spring 2020 concept of homeostasis and regulation: genomics exemplar case study (slos directions: please. Skip to document. University; High School. ... Directions: Please answer each item using short phrases or bullet points. Single spaced lines are acceptable.
Obstructive male infertility. Cystic Fibrosis Case Study. What is cystic fibrosis? Click the card to flip 👆. -Most common fatal genetic disease in the US. -Affects 1/2000 Caucasians of European descent (1/25 are heterozygous) -90% of CF patients have a 3 bp deletion that removes Phe 508. -Missing 1 amino acid. Click the card to flip 👆.
Case Study - Cystic Fibrosis. This case study explores the relationship between the cell membrane and breathing difficulties that occur as a result of the genetic disorder cystic fibrosis. Students look at specific channel proteins in the cell membrane that affect the movement of chloride ions. Different mutations result in different problems ...
This case study is a follow-up to the Cystic Fibrosis Case Study where students explore how changes in transport proteins affects the movement of ions, resulting in a build-up of chloride ions and the symptoms of the disease.. Students were introduced to the idea that different mutations can cause differences in the transport proteins, but in the first version, the origin of these mutations ...
1. Introduction. Cystic fibrosis (CF) is a serious and life-shortening genetic disorder affecting approximately 70,000 persons worldwide [].Respiratory failure is the foremost cause of death in CF patients, and lung transplantation is often considered in end-stage CF disease.
Name(s)Riley_Ferrell_____ Date_____ A Case of Cystic Fibrosis Dr. Weyland examined a six month old infant that had been admitted to University Hospital earlier in the day. The baby's parents had brought young Zoey to the emergency room because she had been suffering from a chronic cough. In addition, they said that Zoey sometimes would "wheeze" a lot more than they thought was ...
Slow growth due to cystic fibrosis the child appears to be having difficulty breathing. nail beds with a bluish hue, with pronounced clubbing can be an indication of hypoxia - often occurs in cystic fibrosis. Pale, warm moist forehead could be a symptom of the child's fever and difficulty breathing sinus tachycardia 160bpm
In addition, many states have introduced newborn screening for CF, resulting in the detection of asymptomatic infants with CF. Case 12. Failure to Thrive: Workup Results in Diagnosis of Cystic Fibrosis. Mr. and Mrs. M, a white couple, have two children, a four-year-old son and a three-month-old daughter. The three-month-old has had considerable ...
This case introduces students to "Maggie," who has just been diagnosed with cystic fibrosis. The students must identify the mutation causing Maggie's disease by transcribing and translating a portion of the wildtype and mutated CFTR gene. Students then compare the three-dimensional structures of the resulting proteins to better understand ...
Case Study - Cystic Fibrosis. Get a hint. What type of genetic disorder is CF and what is the pattern of inheritance? Click the card to flip 👆. autosomal recessive. single-point mutation. the common form is a missense mutation on the 508 position of the amino acid in the CFTR gene [cystic fibrosis transport regulator] Click the card to flip ...
This case study asks students to examine a case of cystic fibrosis. As students read the symptoms and gather evidence about membrane proteins, they learn that CF is really a disorder of membrane permeability. ... Imagine a door with key and combination locks on both sides, back and front. Now imagine trying to unlock that door blind-folded ...
Contributed by Karen Klyczek, University of Wisconsin - River Falls. Mutation/techniques illustrated. For Case A: The DNA sequence files are from the human MP6d9 gene, GenBank accession #M37523, 383 bp, which is linked to the cystic fibrosis gene on chromosome 7. This simulates a PCR-amplified region near the CF gene. The mutated sequence files were created by replacing the G to A base ...
Ap Bio Case Study Assignment case of cystic fibrosis dr. weyland examined six month old infant that had been admitted to university hospital earlier in the day. ... Justify your answer. Part II: CF is a disorder of the cell membrane. Imagine a door with key and combination locks on both sides, back and front. Now imagine trying to unlock that
HESI Cystic fibrosis Case Study Darla. Teacher 25 terms. Debbie_Rowan. Preview. Cystic Fibrosis Case Study. 28 terms. coxcyb. Preview. Objective 3 Child Dev Birth to 5 Set 4. Teacher 7 terms. ageast7. Preview. NCLEX Exam 1. 186 terms. Awesomeunicorn_76. Preview. Sickle Cell Anemia Hesi Case Study. 29 terms. meornela. Preview. Infant Reflexes ...
Answers to the case study: 1. "Woe to that child which when kissed on the forehead tastes salty. He is bewitched and soon will ... In people who have cystic fibrosis, the salt travels to the skin's surface with the water and is not reabsorbed. The proteins are defective remember, so the chlorine cannot pass through to get back into the skin. ...
7. Codon charts sometimes show abbreviations for amino acids rather than the first three letters. The abbreviation for phenylalanine (phe) is F. The most common form of CF is described as F508Del. Explain why this letter combination F508Del is used to describe this particular mutation. Missense Mutations 8. CFTR G551D Mutations - This mutation occurs in about 5% of CF patients.
Cystic fibrosis transmembrane conductance regulator (CFTR) is an ATP-gated anion channel with two remarkable distinctions. First, it is the only ATP-binding cassette (ABC) transporter that is known to be an ion channel—almost all others function as transport ATPases. Second, CFTR is the only ligand-gated channel that consumes its ligand (ATP ...
the CFTR protein is a growth factor receptor four don the surface of lung cells. False; the CFTR protein is a membrane protein responsible for controlling the movement of chloride in and out of cells. in which organelle (s) does transcription occur in eukaryotes. nucleus. in prokaryotes, translation happens after transcription and RNA ...